 La Sindrome di Hutchinson-Gilford( Progeria o HCGP) è una malattia genetica rara e fatale caratterizzata dall’insorgenza di un invecchiamento precoce in età infantile. Il suo nome deriva dal greco e significa “invecchiamento precoce”. Esistono diverse forme di Progeria e la più comune è la Sindrome di Hutchinson-Gilford, dai nomi dei medici che l’hanno descritta per primi in Inghilterra (il dottor Jonathan Hutchinson nel 1886 e il dottor Hastings Gilford nel 1897). L’ HGPS, la forma classica di Progeria, è causata da una mutazione genetica nel gene LMNA che si verifica durante la formazione degli spermatozoi, quindi è di origine paterna, come molte mutazioni spontanee. La ragione è attribuibile al grande numero di divisioni necessarie che le cellule progenitrici fanno per diventare spermatozoi maturi.
La Sindrome di Hutchinson-Gilford( Progeria o HCGP) è una malattia genetica rara e fatale caratterizzata dall’insorgenza di un invecchiamento precoce in età infantile. Il suo nome deriva dal greco e significa “invecchiamento precoce”. Esistono diverse forme di Progeria e la più comune è la Sindrome di Hutchinson-Gilford, dai nomi dei medici che l’hanno descritta per primi in Inghilterra (il dottor Jonathan Hutchinson nel 1886 e il dottor Hastings Gilford nel 1897). L’ HGPS, la forma classica di Progeria, è causata da una mutazione genetica nel gene LMNA che si verifica durante la formazione degli spermatozoi, quindi è di origine paterna, come molte mutazioni spontanee. La ragione è attribuibile al grande numero di divisioni necessarie che le cellule progenitrici fanno per diventare spermatozoi maturi.
Il gene LMNA produce una proteina che si chiama Lamina e che si trova al di sotto della membrana nucleare, formando una sorta di reticolo strutturale dove trovano ancoraggio molte proteine diverse della membrana stessa e i cromosomi. La mutazione porta alla formazione di una lamina più corta (progerina) che diventa tossica per le cellule rendendolo più sensibili ai danni esterni e alterando numerosi collegamenti interni dal punto di vista funzionale.

La Fondazione per la Ricerca sulla Progeria (PFR) ha avuto un ruolo fondamentale nella scoperta del gene responsabile della Progeria riportando, nell’aprile 2003, sul prestigioso giornale scientifico Nature, la scoperta del gene responsabile della Progeria, avvenuta grazie allo straordinario lavoro di un team di scienziati del Genetics Consortium della PRF. Secondo i ricercatori, il processo di invecchiamento precoce tipico della Progeria dipenderebbe proprio da un difetto della Lamina A, che rende il nucleo instabile, provocando instabilità cellulare.
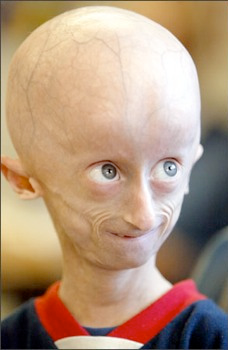
I sintomi della Progeria assomigliano fortemente al normale invecchiamento umano. ma si verificano in bambini piccoli. Essa colpisce un neonato ogni 4-8 milioni, si presenta in tutte le etnie e colpisce indifferentemente maschi e femmine. I bimbi affetti da progeria che sono apparentemente sani alla nascita, già dal primo anno di vita mostrano i primi segni della malattia.
Tra i principali sintomi:
- mancata crescita durante il primo anno di vita,
- calvizie,
- perdita di ciglia e sopracciglia,
- perdita di peso,
- rigidità articolare,
- macrocefalia ( grande testa rispetto alle dimensioni del viso),
- pelle secca, squamosa, sottile,
- viso rattrappito e spiegazzato,
- lussazione delle anche,
- aterosclerosi generalizzata,
- formazione ritardata o assente di denti,
- piccola mandibola( micrognazia)
 I bimbi colpiti da Progeria sono geneticamente predisposti all’insorgenza progressiva e prematura di malattie cardiocircolatorie e il loro decesso è dovuto quasi sempre a disturbi cardiovascolari diffusi. Soffrendo di malattie cardiovascolari, presentano: pressione alta, insorgenza di ictus, angina (dolore al petto dovuto ad un ridotto afflusso di sangue al cuore), ingrossamento del cuore e infarto.
I bimbi colpiti da Progeria sono geneticamente predisposti all’insorgenza progressiva e prematura di malattie cardiocircolatorie e il loro decesso è dovuto quasi sempre a disturbi cardiovascolari diffusi. Soffrendo di malattie cardiovascolari, presentano: pressione alta, insorgenza di ictus, angina (dolore al petto dovuto ad un ridotto afflusso di sangue al cuore), ingrossamento del cuore e infarto.
Di solito la progeria non viene tramandata attraverso le famiglie e raramente si è visto più di un bambino in una famiglia. I pazienti di solito vivono solo fino all’adolescenza, anche se alcuni riescono a raggiungere i 20 anni. L’HGPS è caratterizzata da una mutazione autosomica dominante (perchè è sufficiente solo la mutazione di una copia del gene per il suo manifestarsi) sporadica( perchè è una nuova mutazione).
I bambini colpiti da Progeria invecchiano 8 volte più velocemente del normale: in pratica, sono creature il cui corpo è intrappolato in quello di un anziano, pur con uno sviluppo mentale corrispondente all’età anagrafica, con intelligenza ed emozioni legate ad esso.
Per i genitori che non abbiano mai avuto un bambino con Progeria, la possibilità di mettere al mondo un bimbo con HGPS è di 1 su 4-8 milioni, mentre nel caso in cui abbiano già avuto un bambino con Progeria, il rischio che riaccada, è circa del 2-3%. Questo aumento della percentuale di rischio è legato al mosaicismo, una condizione in cui un genitore ha la mutazione genetica della Progeria in una piccola percentuale delle sue cellule, ma non ha la Progeria.
Dato che il gene della mutazione responsabile della Progeria è stato identificato, la Fondazione per la Ricerca sulla Progeria (PRF) ha in programma un Test diagnostico con cui identificare il cambiamento, la mutazione del gene che causa la sindrome HGPS. Dopo la visita medica e la raccolta dell’anamnesi viene analizzato un campione di sangue per cercare il gene della Progeria, con una precisione scientifica ed in modo precoce, in modo da poter curare i bambini in modo adatto. Non esistono per ora rimedi risolutivi per la progeria ma soltanto trattamenti per curare o prevenire alcune conseguenze provocate dalla malattia come i rischi vascolari, l’ictus, l’infarto e le cardiopatie. La ricerca sta però tentando di invertire il processo genetico, cercando di limitare i danni della tossicità della progerina mediante l’impiego di inibitori specifici che la rendono più instabile e quindi più facilmente eliminabile. Attualmente esistono protocolli sperimentali diversi utilizzati in Francia, Stati Uniti e Italia che riguardano l’impiego combinato di farmaci già noti per altre malattie: la Pravastatina e Acido Zoledronico.
Questi farmaci agiscono sulla via di formazione della lamina (progerina) matura e quindi interferiscono con la sua modificazione chimica (farnesilazione), rendendo la proteina meno tossica. Altri studi hanno utilizzato un farmaco sperimentale, il Lonafarnib, che sarebbe in grado di bloccare alcuni degli effetti devastanti della proteina progerina: in tutti i bambini trattati con questo farmaco, sono stati registrati degli effetti positivi, come un aumento di forza muscolare o un miglioramento della condizione dei vasi sanguigni.
 Vuoi ricevere le notifiche sulle nostre notizie più importanti?
Vuoi ricevere le notifiche sulle nostre notizie più importanti?{kind=link}