Gli occhi del mondo della medicina sono puntati in queste settimane sui casi di vaiolo delle scimmie riscontrati nell’uomo in diversi Paesi del mondo. La macchina scientifica è al lavoro per cercare di ottenere informazioni su questo virus il più rapidamente possibile, in modo da poter adottare le giuste misure. Finora l’invito generale è quello di evitare allarmismi ingiustificati. È lo stesso messaggio comunicato dal biologo Enrico Bucci, Adjunct Professor presso la Temple University di Philadelphia. In due diversi post su Facebook, Bucci parla degli ultimi aggiornamenti emersi dal sequenziamento del virus responsabile dei casi attuali.
“Questa mattina risultavano coinvolte 17 nazioni al di fuori dell’Africa, tra cui per ultima l’Argentina, per un totale di 194 segnalazioni, di cui una risultata un falso allarme (in Israele), 107 confermate e le restanti sotto indagine”, scrive Bucci, spiegando che “genere dei contagiati, loro sintomi e ospedalizzazioni non cambiano significativamente”.
“Il consorzio Nextstrain ha cominciato ad analizzare le sequenze disponibili di MPX; in particolare, i ricercatori si sono concentrati sull’analisi di 29 genomi appartenenti al raggruppamento che si è originato nell’Africa occidentale, cui appartengono le tre sequenze sin qui disponibili per i casi odierni”, continua Bucci. Nella figura in basso, “ogni cerchietto rappresenta un gruppo di sequenze in un Paese, il cui diametro è proporzionale al numero di campioni sequenziati; in giallo, due dei tre campioni attuali, in celeste quelli isolati dal 1968 sino ad oggi. Le linee rosse uniscono Paesi che presentano almeno due genomi sostanzialmente identici fra loro, e quindi mostrano le presunte rotte di diffusione del contagio. Come vedete, sono decenni che si osserva la diffusione di questo particolare ceppo dell’Africa occidentale, e i Paesi coinvolti non sono poi molto diversi da quelli odierni; da quando è noto, questo ceppo è risultato trasmettersi da uomo a uomo e su lunga distanza al di fuori dell’Africa, contrariamente a ciò che certi articolisti in cerca di risonanza scrivono”, si legge nel post.
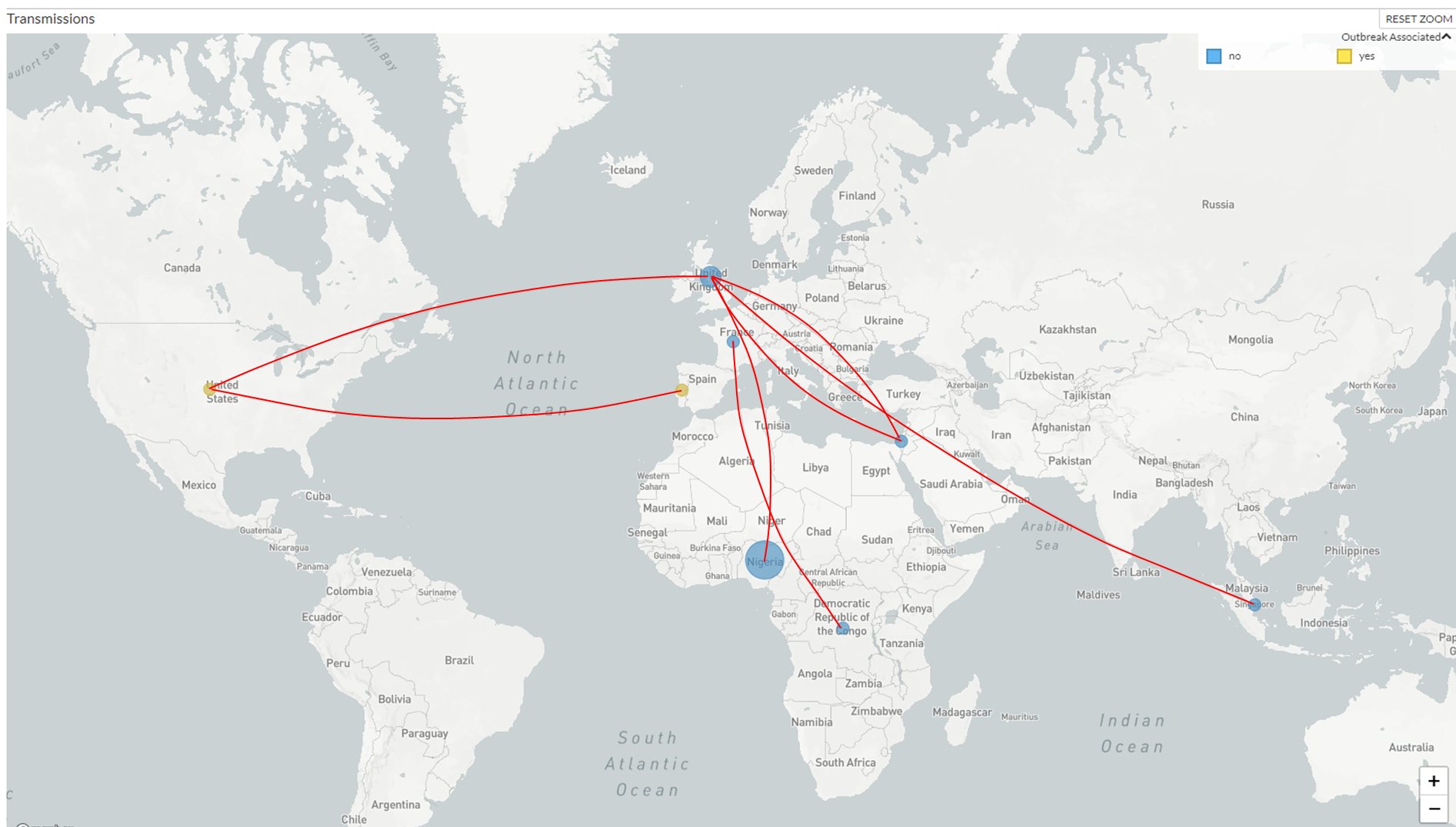
“Ogni anno, in Nigeria si registrano circa 3000 casi, con prevalenza in giovani maschi come osserviamo oggi, e questi sono solo quelli noti; più volte negli ultimi decenni, inoltre, si sono avuti outbreak da svariate centinaia di casi nel mondo, e le linee di connessione fra i genomi isolati in Paesi diversi mostrano che si tratta di una sorta di “ribollire” continuo che va avanti da un bel pezzo. Insomma: stiamo osservando l’ultimo fotogramma di un film cominciato almeno 50 anni fa, con un virus che – come ha sostenuto Roberto Burioni – è un virus “vecchio”. Non è il caso di preoccuparsi troppo, e bisogna invece attendere i dati. Come sempre, adesso è il momento di studiare e approfondire bene nei laboratori, non quello di lanciare allarmi; come sempre, insomma, sereni ma vigili”, continua il biologo.
“I dati cominciano ad arrivare numerosi dai laboratori di tutto il mondo, perché la comunità scientifica sta lavorando anche questa volta a tempo di record. I ricercatori portoghesi, in particolare, hanno rilasciato la sequenza di altri 9 isolati virali, in aggiunta a quello originale; così disponiamo adesso di 10 genomi dal Portogallo, uno dagli USA e uno dal Belgio. Quello belga per ora lasciamolo da parte, perché sono emersi problemini tecnici nel sequenziamento”, spiega Bucci in un altro post.
“Analizzando il fantastico lavoro fatto dai ricercatori dello INSA di Lisbona, con i dati subito messi a disposizione di tutti, ora possiamo fare le seguenti considerazioni.
1) Si conferma l’origine di tutti i virus sin qui sequenziati dal ceppo dell’Africa Occidentale, meno pericoloso.
2) E’ probabile che l’origine dell’attuale outbreak sia in un singolo “caso zero”, almeno per quel che riguarda gli 11 genomi da USA e Portogallo; le sequenze sono davvero molto simili.
3) La differenza fra i virus attuali e quello più simile del 2018 è in almeno 46 diverse posizioni del genoma, e la differenza media dai virus del corrispondente outbreak del 2018 è in circa 50 posizioni. Si tratta di una differenza maggiore di quanto atteso, sulla base della velocità di mutazione nota per questi virus sinora.
4) Come messo in evidenza da Andrew Rambaut, dell’Università di Edimburgo, per spiegare questa maggior velocità di mutazione, possiamo considerare che quasi tutte le mutazioni riscontrate sono di un tipo caratteristico della risposta dell’ospite infetto (umano o animale) all’infezione del virus, attraverso un enzima chiamato APOBEC3, che ipermuta i genomi invasori per renderli non funzionanti, ma può generare occasionalmente nuovi virus mutanti.
5) All’interno del gruppo di virus corrispondenti all’odierno outbreak, si cominciano a notare piccoli raggruppamenti, che suggeriscono una microevoluzione ancora in corso”, conclude Bucci.
 Vuoi ricevere le notifiche sulle nostre notizie più importanti?
Vuoi ricevere le notifiche sulle nostre notizie più importanti?